存在しないウイルスの出現を予測したAI、ウイルス進化の未来を読み解く『CoVFit』の実力
【研究って楽しい】東京大学医科学研究所・伊東潤平准教授が挑むウイルスの次期流行株予測(後編)
2025.5.6(火)
ここからは、JBpress Premium 限定です。
カンタン登録!続けて記事をお読みください。詳細はこちら



あわせてお読みください

折り紙でホイップクリームを作る?独自開発のソフトウェアと折り紙で無限のかたちを作り出す教授の突き抜けた発想
【研究って楽しい】折り紙をサイエンスにした筑波大学システム情報系・三谷純教授が語る、奥深すぎる折り紙の世界
関 瑶子

ウイルスの変化は予測できるか?変異株の「適応度」がカギを握る、次のパンデミックを防ぐ科学の最前線
【研究って楽しい】東京大学医科学研究所・伊東潤平准教授が挑むウイルスの次期流行株予測(前編)
関 瑶子

古代人のウンコの化石からこんなことまでわかる! 新たな学問「バイオインフォマティクス」の底力
【研究って楽しい】東京大学・医科学研究所の西村瑠佳が語る「生物学×情報科学」の魅力
関 瑶子

46億年前の地球に大陸はなかった!大陸はいつ、どのようにして誕生したのか?気鋭の岩石学者が挑む大陸誕生の謎
【著者が語る】『大陸の誕生』の田村芳彦氏が語る岩石学の魅力と、地震学との関係、岩石学から見た能登半島地震
関 瑶子

【物理学超入門】「光は波か粒子か」からスタートした量子力学、その歴史を物理学者の野村泰紀が語る
【著者に聞く】ホイヘンスにヤング、マクスウェル、プランク、ボーア、そしてシュレディンガー、続々と登場する物理界のスター
関 瑶子
本日の新着

AIとの「協働」が突きつける新たなジレンマ―ハーバード大が描く「サイバネティック・チームメイト」の光と影
生産性向上も「多様性の喪失」と「育成の空洞化」が壁に
小久保 重信

韓国の一大社会問題へ発展した「注射おばさん」と「点滴おばさん」事件
医師免許を持たず規制薬物を芸能人に日常的投与か
アン・ヨンヒ

でっち上げた疑惑でパウエル議長を刑事捜査、トランプの狙いは「FRBの隷属」、中央銀行の独立性をいとも簡単に蹂躙
木村 正人

【関連銘柄も爆上がり】2035年に6兆円市場に、AI業界が注力するフィジカルAI、日本はロボット大国の地位を守れるか
【生成AI事件簿】変わるゲームのルール、日本が乗り越えなければならない5つの壁と、日本が取るべき3つの戦略
小林 啓倫
明日の医療 バックナンバー

来年開始の医学部定員削減で「地域医療崩壊」に現実味、この課題に医学部受験専門予備校「京都医塾」が取り組む理由
三重 綾子

【アルコールは発がん物質】アルコールの毒は一種類ではない、細胞の「傷つけ役」が体内で次々に増えていく怖さ
齊藤 康弘

【高額療養費制度見直し】「財源に限りが、だから困っている人だけ助ける」では救われない重病・難病患者が続出する
坂元 希美

身体がほとんど動かない重度障害の患者との意思疎通は可能か?微かな動きから読み取る人々と動かない身体が語ること
長野 光 | 西村 ユミ
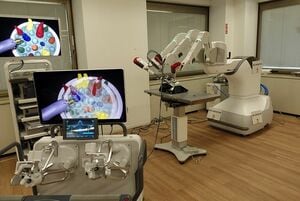
ワンちゃんネコちゃん向けのAIロボット手術が登場、航空機のパイロットのように獣医師が自動操縦で手術も
星 良孝
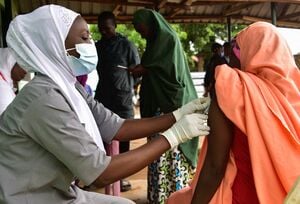
エムポックス1bが日本初確認、WHOが緊急事態を解除した直後に走る緊張、今の状況はどうなっている?
星 良孝
フォロー機能とは、指定した著者の新着記事の通知を受け取れる機能です。
フォローした著者の新着記事があるとヘッダー(ページ上部)のフォロー記事アイコンに赤丸で通知されます。![]()
フォローした著者の一覧はマイページで確認できます。![]()
※フォロー機能は無料会員と有料会員の方のみ使用可能な機能です。
設定方法
記事ページのタイトル下にある「フォローする」アイコンをクリックするとその記事の著者をフォローできます。
確認方法
フォロー中の著者を確認したい場合、ヘッダーのマイページアイコンからマイページを開くことで確認できます。
解除方法
フォローを解除する際は、マイページのフォロー中の著者一覧から「フォロー中」アイコンをクリック、
または解除したい著者の記事を開き、タイトル下にある「フォロー中」アイコンをクリックすることで解除できます。